 W
WZespół Angelmana, AS – genetycznie uwarunkowany zespół spowodowany najczęściej mikrodelecją w regionie 15q11-13.
 W
WZespół Cornelii de Lange – grupa zespołów wad wrodzonych uwarunkowanych genetycznie, o możliwym różnym typie dziedziczenia, których wspólną cechą jest współistnienie następujących objawów:niepełnosprawność intelektualna opóźnienie wzrastania i w wyniku tego niski wzrost krótkogłowie niskie osadzenie uszu szyja z podłużnym fałdem skórnym przechodząca na klatkę piersiową zrośnięte brwi (synophrys), które są bardzo wyraźne, jakby narysowane ołówkiem, zwykle z długimi rzęsami długa warga górna, kąciki ust skierowane są ku przodowi, podobnie jak nozdrza, podbródek jest mały wady kończyn – płaskie, małe dłonie i stopy, czasami występuje brak palców częste wady serca refluks żołądkowo-przełykowy głuchota.
 W
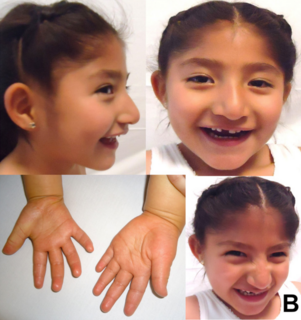
WZespół Costello – genetycznie uwarunkowany zespół wad wrodzonych, dziedziczony autosomalnie dominująco. Charakteryzuje się niedoborem wzrostu, opóźnionym rozwojem psychoruchowym, nadmierną wiotkością małych stawów, hiperpigmentacją skóry, charakterystycznym wyglądem twarzy oraz niepełnosprawnością intelektualną.
 W
WZespół Downa, dawniej nazywany mongolizmem – zespół wad wrodzonych spowodowany obecnością dodatkowego materiału genetycznego chromosomu 21. Eponim pochodzi od nazwiska brytyjskiego lekarza Johna Langdona Downa, który opisał go w 1866 roku. Występowanie trisomii chromosomu 21 opisane zostało w 1959 r.
 W
WZespół Dubowitza – genetycznie uwarunkowany zespół wad wrodzonych charakteryzujący się niepełnosprawnością intelektualną, niedoborem wzrostu oraz zaburzeniami odporności predysponującymi do alergii, wyprysku, nowotworów układu krwiotwórczego oraz nerwiaka zarodkowego.
 W
WZespół Edwardsa – zespół wad wrodzonych spowodowany trisomią chromosomu 18. Częstość występowania zespołu szacowana jest na 1:8000 urodzeń. We wczesnym okresie rozwoju zarodka częstość wystąpienia trisomii 18 jest większa niż w późniejszym okresie rozwoju. 95% płodów dotkniętych tą wadą ulega poronieniu. Rodzi się więcej noworodków żeńskich, co może wynikać z faktu, że płody męskie z trisomią chromosomu 18 są ronione częściej. Dodatkowa kopia chromosomu 18 powoduje przyrost masy DNA o 2,8%.
 W
WZespół Johanson-Blizzarda – genetycznie uwarunkowany zespół wad wrodzonych charakteryzujący się niedoczynnością zewnątrzwydzielniczą trzustki, hipoplazją skrzydełek nosa, hipodoncją, niedosłuchem odbiorczym oraz niepełnosprawnością intelektualną.
 W
WZespół Kabuki typu 2 – rzadki zespół wad wrodzonych związany z niepełnosprawnością intelektualną. Został opisany po raz pierwszy u dziesięciorga pacjentów przez Norio Niikawę i Yoshikazu Kurokiego i ich współpracowników w 1981 roku. Nazwa zespołu odzwierciedla charakterystyczny wygląd twarzy pacjentów z tym zespołem, przypominający ucharakteryzowane twarze aktorów tradycyjnego japońskiego teatru Kabuki (Kumadori).
 W
WOgniskowa dysplazja korowa (ang. focal cortical dysplasia, FCD) - wada w budowie tkanki nerwowej i dojrzewania komórek w obrębie kory mózgowej, nie jest to zmiana nowotworowa. Powstaje w wyniku zaburzenia migracji i różnicowania komórek mózgowych. Zarówno czynniki genetyczne, jak i pozagenetyczne biorą udział w jej powstaniu, dokładna etiologia nie została jednak poznana. Przypuszcza się, że mutacje w genie "TSC1" mogą mieć udział w powstaniu FCD.
 W
WZespół Pallistera-Killiana in. PKS – genetycznie uwarunkowany zespół wad wrodzonych, w którym aberracja chromosomowa występuje w postaci mozaiki ograniczonej do niektórych tkanek. Charakteryzuje się dysmorfią twarzy, zaburzeniami pigmentacji skóry, alopecją, częstym występowaniem wad przepony oraz dodatkowych brodawek sutkowych wraz niepełnosprawnością intelektualną oraz padaczką.
 W
WZespół Pradera-Williego – zespół wad wrodzonych spowodowany aberracją chromosomalną, najczęściej częściową utratą (delecją) długiego ramienia chromosomu 15, pochodzącego od ojca. Na obraz kliniczny choroby składają się niski wzrost, upośledzenie umysłowe, niedorozwój narządów płciowych (hipogonadyzm) oraz otyłość spowodowana mniejszym niż u zdrowych ludzi zapotrzebowaniem energetycznym przy jednoczesnym ciągłym niepohamowanym uczuciu głodu. Uważa się, że zespół Pradera-Williego jest najczęstszą genetycznie uwarunkowaną przyczyną otyłości.
 W
WZespół Retta – neurologiczne zaburzenie rozwoju, uwarunkowane genetycznie, o dziedziczeniu sprzężonym z płcią.
 W
WStwardnienie guzowate, choroba Bourneville’a-Pringle’a, TSC – rzadka, wielonarządowa choroba uwarunkowana genetycznie, należąca do grupy fakomatoz, powodująca zmiany w nerkach, sercu, gałkach ocznych, mózgu, płucach i skórze, często związana z niepełnosprawnością intelektualną i padaczką. Stwardnienie guzowate zostało opisane jako nowa jednostka chorobowa w 1880 roku przez Désiréa-Magloire’a Bourneville’a. Przez ostatnie dwa dziesięciolecia dokonał się znaczny postęp w wiedzy o tej chorobie, związany z odkryciem jej genetycznego podłoża – mutacji w genach TSC1 i TSC2. Stwarza on nowe możliwości skutecznej terapii stwardnienia guzowatego.
 W
WZespół Williamsa – rzadki, genetycznie uwarunkowany zespół wad wrodzonych. Opisał go w 1961 roku nowozelandzki kardiolog J. C. P. Williams.
 W
WZespół Wolfa-Hirschhorna – zespół wad wrodzonych spowodowany mikrodelecją na krótkim ramieniu chromosomu 4. Około 20% dzieci chorych na ten zespół nie dożywa 2 roku życia. Częstość zespołu szacuje się na około 1:50 000 urodzeń; występuje częściej u dziewczynek.
 W
WZespół łamliwego chromosomu X, zespół kruchego chromosomu X, zespół Martina-Bell – choroba genetyczna cechująca się obniżeniem poziomu rozwoju intelektualnego różnego stopnia, i której niektóre objawy behawioralne niekiedy pokrywają się z objawami charakterystycznymi dla autyzmu.